Pharmacology - Tumblr Posts

I got a more than perfect score on my pharmacology exam! I’m feeling better and better about prescribing! #np #nursepractitioner #nursingstudent #npstudent #pharmacology #school #goodgrades #gradschool (at San Diego, California) https://www.instagram.com/p/BpX42uJgA3f/?utm_source=ig_tumblr_share&igshid=fd0xvglhcid8
Pharmacology

When pharmac is also laughing at your pitiable condition

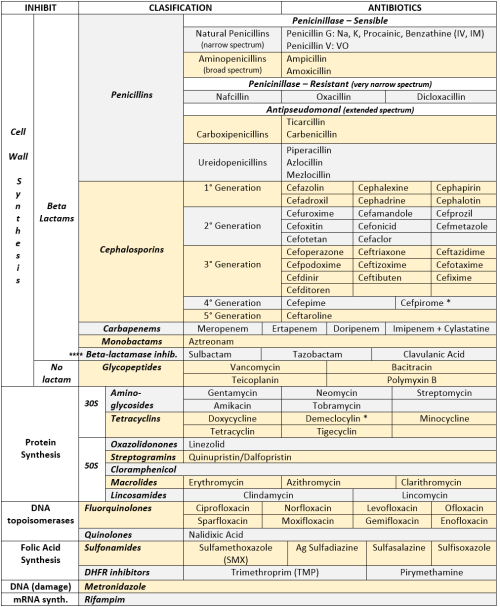
ANTIBIOTICS CHEAT SHEET :)
Also, REMEMBER!!!!
* Sulfonamides compete for albumin with:
Bilirrubin: given in 2°,3°T, high risk or indirect hyperBb and kernicterus in premies
Warfarin: increases toxicity: bleeding
* Beta-lactamase (penicinillase) Suceptible:
Natural Penicillins (G, V, F, K)
Aminopenicillins (Amoxicillin, Ampicillin)
Antipseudomonal Penicillins (Ticarcillin, Piperacillin)
* Beta-lactamase (penicinillase) Resistant:
Oxacillin, Nafcillin, Dicloxacillin
3°G, 4°G Cephalosporins
Carbapenems
Monobactams
Beta-lactamase inhibitors
* Penicillins enhanced with:
Clavulanic acid & Sulbactam (both are suicide inhibitors, they inhibit beta-lactamase)
Aminoglycosides (against enterococcus and psedomonas)
* Aminoglycosides enhanced with Aztreonam
* Penicillins: renal clearance EXCEPT Oxacillin & Nafcillin (bile)
* Cephalosporines: renal clearance EXCEPT Cefoperazone & Cefrtriaxone (bile)
* Both inhibited by Probenecid during tubular secretion.
* 2°G Cephalosporines: none cross BBB except Cefuroxime
* 3°G Cephalosporines: all cross BBB except Cefoperazone bc is highly highly lipid soluble, so is protein bound in plasma, therefore it doesn’t cross BBB.
* Cephalosporines are "LAME“ bc they do not cover this organisms
L isteria monocytogenes
A typicals (Mycoplasma, Chlamydia)
M RSA (except Ceftaroline, 5°G)
E nterococci

* Disulfiram-like effect: Cefotetan & Cefoperazone (mnemonic)
* Cefoperanzone: all the exceptions!!!
All 3°G cephalosporins cross the BBB except Cefoperazone.
All cephalosporins are renal cleared, except Cefoperazone.
Disulfiram-like effect
* Against Pseudomonas:
3°G Cef taz idime (taz taz taz taz)
4°G Cefepime, Cefpirome (not available in the USA)
Antipseudomonal penicillins
Aminoglycosides (synergy with beta-lactams)
Aztreonam (pseudomonal sepsis)
* Covers MRSA: Ceftaroline (rhymes w/ Caroline, Caroline the 5°G Ceph), Vancomycin, Daptomycin, Linezolid, Tigecycline.
* Covers VRSA: Linezolid, Dalfopristin/Quinupristin
* Aminoglycosides: decrease release of ACh in synapse and act as a Neuromuscular blocker, this is why it enhances effects of muscle relaxants.
* DEMECLOCYCLINE: tetracycline that’s not used as an AB, it is used as tx of SIADH to cause Nephrogenic Diabetes Insipidus (inhibits the V2 receptor in collecting ducts)
* Phototoxicity: Q ue S T ion?
Q uinolones
Sulfonamides
T etracyclines

* p450 inhibitors: Cloramphenicol, Macrolides (except Azithromycin), Sulfonamides
* Macrolides SE: Motilin stimulation, QT prolongation, reversible deafness, eosinophilia, cholestatic hepatitis
* Bactericidal: beta-lactams (penicillins, cephalosporins, monobactams, carbapenems), aminoglycosides, fluorquinolones, metronidazole.
* Baceriostatic: tetracyclins, streptogramins, chloramphenicol, lincosamides, oxazolidonones, macrolides, sulfonamides, DHFR inhibitors.
* Pseudomembranous colitis: Ampicillin, Amoxicillin, Clindamycin, Lincomycin.
* QT prolongation: macrolides, sometimes fluoroquinolones

Molecule of the Day: Methylphenidate


Methylphenidate (C14H19NO2), also known as Ritalin, is a white powder that is slightly soluble in water. It is commonly used to treat ADD, ADHD, and narcolepsy.
Methylphenidate inhibits dopamine and norepinephrine transporter proteins, thus preventing dopamine and norepinephrine in the synaptic cleft from being reuptaken into the presynaptic knob. The resultant higher concentration of these substances in the synapse causes the receptors on the postsynaptic knob to be stimulated at a greater frequency, thus achieving greater synaptic transmission. This produces a psychostimulant effect, allowing it to be used in the treatment of ADHD.

In small amounts, methylphenidate has also been shown to enhance memory and control, caused by the activation of dopamine and adrenergic receptors. However, in large doses, it can have the opposite effect.
It has few side effects, which include loss of appetite, nausea, and insomnia. However, like many strong dopamine reuptake inhibitors, it can result in dependence, and is often seen as a gateway drug.
Methylphenidate is industrially synthesised through a multi-step pathway from 2-bromopyridine and benzyl cyanide.

Requested by @zenbra
Molecule of the Day: Diazepam/Valium


Diazepam (C16H13ClN2O), also known as Valium, is a white solid that is of significant pharmaceutical importance. It is a member of the benzodiazepine family, which shares the similar bicyclic system comprising of a conjoined benzene and diazepine ring.
Diazepam is used to treat anxiety and panic disorders, and this is achieved by its binding to GABA receptors on neurons. This causes the active site of the receptors to become a better fit for GABA molecules, resulting in a higher binding of GABA to it. This triggers a greater influx of chloride ions into the neuron.

Since the intracellular portion of the neuron is more negative than normal, the membrane is hyperpolarised to a greater extent. Consequently, a stronger stimulus is needed to trigger an action potential, which is created when a stimulus causes the membrane to reach the threshold potential.
Since the resting potential is now more negative, the action potential and thus firing of the neuron is less likely. This then produces the anxiolytic, sedative, amnesia-inducing, and anticonvulsant effects of diazepam.

Diazepam can be produced by various synthetic pathways; one such one is shown below.

Requested by anonymous
Grapefruit Juice & Drug Metabolism
Bergamottin is a natural furanocoumarin found principally in grapefruit juice. It is also found in the oil of bergamot, from which it was first isolated and from which its name is derived.
To a lesser extent, bergamottin is also present in the essential oils of other citrus fruits. Along with the chemically related compound 6’,7’-dihydroxybergamottin, it is believed to be responsible for the grapefruit juice effect in which the consumption of the juice affects the metabolism of a variety of pharmaceutical drugs.

Pharmacokinetics Overview
(Absorption and distribution of drugs)
The study of the time course of drugs and their metabolites in the body (what the body does to the drug) consisting of:
administration
absorption
distribution
metabolism
excretion
Administration
Enteral (passes through intestine)
oral (mouth)
buccal/sublingual (applied in cheek/under tongue)
Gastrosomy (surgical opening through the abdomen into the stomach)
Topical (applied directly)
Nasal
Rectal
Ophthalmic (eyes)
Parentral (injection)
Intravenous (into veins)
intramuscular (into muscles)
intradermal (within layers of skin)
subcutaneous (under the skin)
Drug molecules move around the body either through bulk flow (bloodstream, lymphatics or cerebrospinal fluid) or diffusion (molecule by molecule over short distances)

Absorption
Passage of drug from its site of administration into plasma - important for all routes except intravenous injection.
Injection
IV = fastest route of administration
bolus injection = very high concentration of drug
rate limiting factors = diffusion through tissues and removal by local blood flow
Drugs need to pass through membranes (cell membranes, epithelial barriers, vascular endothelium, blood-brain barrier, placenta barrier etc) via
passive diffusion through lipids
carrier-mediated
passage through membrane pores/ion channels
pinocytosis (ingestion into a cell by the budding of small vesicles from the cell membrane)
Diffusion through lipid
non-polar molecules can dissolve freely in membrane lipids
the rate is determined by the permeability coefficient (P)(solubility in the membrane and diffusibility) and the concentration difference across the membrane
pH and Ionisation
Many drugs are weak acids or weak bases
exist in unionised or ionised forms
pH = balance between the two forms
ionised forms have low lipid solubility
uncharged however the drug is usually lipid soluble

ionisation affects:
rate of drug permeation through membranes
steady state distribution of drug molecules between aqueous compartments if pH difference exists between them
Therefore:
urinary acidification accelerates the excretion of weak bases and slows that of weak acids
alkalisation has opposite effect
increasing plasma pH causes weak acids to be extracted from CNS into plasma
Reducing plasma pH causes weakly acidic drugs to become concentrated in CNS, increasing neurotoxicity
Bioavailibility
Bioavailibility (F) indicates the fraction of an orally administered dose that reaches systemic circulation intact, taking into account both absorption and local metabolic degradation
determined by comparison between oral and IV absorption

affected by:
drug preparation
variation in enzyme activity of gut
gastric pH
intestinal motility
Volume of Distribution
Vd is defined as the volume of fluid required to contain the total amount, Q, of drug in the body at the same concentration as that present in the plasma, Cp
determined by relative strength of binding between drug and tissue compared with drug and plasma proteins
tight binding to tissue but not plasma –> drug appears to be dissolved in large volume –> large Vd (eg chloropromazine)
tight binding to plasma –> V can be very close to blood volume –> low Vd (eg warfarin)


PHARMACOKINETICS CALCULATIONS
Single-Dose: •Vd = Dose/Co •t1/2 = 0.7 x Vd/Cl
Multiple-Dose, Infusions: •Ko = Cl x Css •LD = Vd x Cp/f •MD = Cl x Css x dosing interval/f
Vd: volume of distribution Co: plasma [ ] at zero time Cp: plasma [ ] t1/2: half life Cl: clearance = free fraction x GFR Ko: infusion rate Css: steady state [ ] LD: loading dose MD: maintaining dose f: bioavailability
Quantitative Structure-Activity Relationship (QSARs) - Drug Development
QSAR modelling predicts the biological activity of a compound based off its physical properties. They are used to avoid synthesising and testing every possible version of a molecule to find the optimum for bioactivity. A small number of structurally similar molecules are synthesised and tested, and these results are used to mathematically predict other similar molecules on a computer.
Hydrophobicity
This dictates the ease at which a molecule will pass through a cell membrane. Too hydrophobic and the molecule will be drawn to lipids and its bioactivity will be reduced, too hydrophilic and the molecule will be too polar to pass through the phospholipid bilayer and will not carry out its desired activity (will be excreted in urine)
LogP - a measure of the whole molecule’s hydrophobicity

High logP = more hydrophobic
Low logP = more hydrophillic (polar)
Optimum for bioavailibility = 2-4.5
A regression equation can be formed with c=concentration for max activity
1/c = K1 logP + k2
If linear, values for other similar structures can be taken off the line. If parabolic = logP^2, indicating that after a max concentration bioavalibility will not increase as the drug becomes too hydrophobic and moves into fats.
Substituent hydrophobicity constant, π
Measures the hydrophobicity of individual substituents in a compound.
π = logPX - logPH
X= partition coefficient for substituted compound
H= partition coefficient for unsubstituted compound (Hydrogen (so if H was in place of the substituent of interest))
Compares how hydrophobic a substituent is compared to hydrogen
π = +ve –> X= more hydrophobic than hydrogen
π = -ve –> X= less hydrophobic than hydrogen
Note: can be used to calculate logP by adding substituents, rather than having to synthesise and test the molecule (clogP = calculated logP)
Electronics
Pharmacokinetics (administration, distribution, metabolism and excretion) rarely depends on hydrophobicity alone. The polarity of a compound dictates its passage through the patient and its binding at point of activity.
Hammett substituent constant, σ
The starting point is a chemical equilibrium for which both the substituent constant and the reaction constant are arbitrarily set to 1: the ionization of benzoic acid (R and R’ both H) in water at 25 °C.

Provides K0.
RCO2H <–> RCO2- + H+
uses the dissociation constant
kH = [RCO2-][H+] / [RCO2H]
If X is electron withdrawing, it will stabilise RCO2- and shift the equilibrium to the right. kX will increase
eg NO2, CN, Cl –> +ve σ
If X is electron donating, it will destabilise the RCO2- anion and shift equilibrium to the left, with a drop in kX.
eg alkyls, ethyls, methyls = -ve σ
σ = logkX - logkH
Steric properties
Taft steric parameter, Es = rate of hydrolysis of XCH2CO2Me under acidic conditions
Es = logkX - logKH
If X is physically small, the rate of hydrolysis (time taken to reach tetrahedral intermediate) will be fast.

Here, the size of R affects the rate of reaction by blocking nucleophilic attack by water.

H 1.24 +ve value: little steric resistance to hydrolysis Me 0.00 the reference substituent in the Taft equation t-Bu -2.78 -ve value: large resistance to hydrolysis
Small X = large Es, large X = small Es
Accuracy of calculation decreases as the bulk and length of the chain increases.

Hansch equations put several of the parameters together to compare overall bioavailibility of different compounds.
Craig plots
Plots 2 constants
functional groups with similar activity will be in the same quadrant
the optimum quadrant, eg +ve σ and -ve π, will contain all the substituents worth investigating

If someone has an 'enhanced metabolism' that processes drugs faster, is it easier or harder for them to overdose? Wouldn't they uptake more of a drug quicker, therefor making it more dangerous? I see this a lot with Captain America and Spider-man and such, where the dose of various medicines will be raised and I'm not sure that makes sense?


…oooooh you sure have opened a can of pharmacokinetic worms here….
Simply put, whether the drug never reaches therapeutic blood levels, or exceeds them, depends on 1) how the character’s metabolism works, and 2) what kind of drug they ingested (skip to the bolded part at the end of the post to get the tl;dr).
When you take a drug, the following happens (this process is sometimes denoted as “ADME” or “LADME”:
The drug must separate from the vehicle that brought it into the body (for example, a pill must disintegrate in the stomach, releasing the drug, or an IM or IV drug must separate from its solution): Liberation
The drug must be absorbed into the bloodstream (for a pill this would mean getting absorbed through the lining of the stomach or intestine, for IM injections this means getting absorbed by blood vessels running through the muscle where the drug is): Absorption
The drug must be deposited from the bloodstream into a location where it can be used: Distribution
The drug must be metabolized (broken down or changed by a biologic process, creating different chemicals called metabolites): Metabolism
The drug metabolites must be excreted from the body: Elimination
The first end of this process is largely driven by regular old chemistry. A pill has to dissolve to release the drug, and assuming that these characters have similar stomach/small intestine environments, this is not going to be different for them.
Absorption is mostly driven by a concentration gradient (substances like to be at the same concentration across membranes, so if there’s more drug in the small intestine than there is in the blood around the small intestine, the drug gets absorbed into the blood as the concentrations try to equalize), so this too is probably not going to be all that different. Even distribution is (mostly) driven by that concentration gradient, so, again, this process wouldn’t necessarily be any different from that of a normal human.
Now, the latter half of this process is a lot more dependent on a person’s specific physiology. When we talk about metabolism, we’re talking about how the body changes ingested chemicals into something excrete-able. For many drugs, this change involves enzymes in the liver.
About 6 different liver enzymes are responsible for the metabolism of about 90% of drugs. Each different enzyme is responsible for the breakdown of a different group of drugs.
Some people have genetic mutations that cause more or less of an enzyme to be produced. People who make more of an enzyme metabolize that group of drugs faster, while people who make less of them metabolize those drugs more slowly.
Through certain genetic tests, real life people can be designated one of the following for any given group of drugs:
Ultrarapid Metabolizers have the genetic wiring to produce way, way more copies of an enzyme than the typical person, and metabolize the corresponding drugs very, very quickly
Extentive Metabolizers produce more copies of the enzyme than most people
Intermediate Metabolizers produce an average number of copies
Poor Metabolizers produce significantly fewer copies than average, leaving them unable to metabolize the drugs normally
Now, remember how I said it also has to do with what kind of drug it is? There are two different kinds of drugs I’m talking about: Active Drugs and Prodrugs. Active drugs are able to be used by the body as-is. Prodrugs only have an effect once they’ve been metabolized by the body into a different substance.
Say someone is an ultrarapid metabolizer of an active drug. They take the drug, it gets absorbed and distributed like normal, but they rapidly metabolize it into inactive substances and excrete it. This person would either get no effect from a typical dose, or only a very slight one, because the drug is never allowed to build up to effective levels in their blood before getting metabolized.
But say that same person is a poor metabolizer of a different active drug. They take the drug, it gets absorbed, but they only very slowly are able to metabolize and excrete it. The drug ends up building up in their blood and staying there longer, possibly causing an overdose of the drug at a typical dose.
The situation would be reversed if the drugs were prodrugs instead. An ultrarapid metabolizer of a prodrug ends up metabolizing too much of the active substance too quickly, possibly causing overdose, while a poor metabolizer of a prodrug maybe never metabolize enough to get effective concentrations of the end substance (check out this post on the prodrug codeine).
Finally, applying this real-life precedent to Captain America, or Flash, or Spider-Man canon evidence, you would have to assume that due to their respective super powers, they all produce a metric sh*tton of liver enzymes capable of metabolizing drugs super fast and hella effective kidneys for excreting them. Typical doses of active drugs would barely work on them, while prodrugs might have a short, but incredibly strong effect on them.
R E F E R E N C E S
Anti-epileptics/Anti-convulsants Made Incredibly Easy
TREATMENT STRATEGIES:
Start therapy after the second seizure; first ONLY if recurrence is high = MRI abnormal, EEG abnormal, or status epilepticus.
Monotherapy until seizures are controlled.
If failed: titrate up to maximum tolerated dose –> shift to alternative drug –> use drug combination –> VNS, DBS.
Full drug therapy for 2 – 3 years after the last fit.
Gradual withdrawal over at least 6 months.
Rx Profile:


(Drawings are courtesy of @mynotes4usmle)
Carbamazepine
Mainly for generalized tonic-clonic seizures
Trigeminal neuralgia
Bipolar disorders (with depressive predomince) - mood stabelizier
NEVER in abscence seizures
SE:

Lamotrigine
Safer profile, with minimal interactions.
Bipolar disorders (with depressive predominance) - mood stabilizer
SE: maculopapular rash; SJS
Topiramate
Broad spectrum anti-seizure; used in migraine.
SE of TopIRamate: enzyme Inhibitor + Renal stones.
TREATMENT PROTOCOL:

Green: first line; Yellow: second line; Orange: third line; Red: contraindications. (Graph reproduced from Oxford Handbook of Clinical Medicine)
Epilepsy & Pregnancy:
Non-enzyme-inducing AEDS have no effect on the pill. Enzyme inhibitors prolong the half life of OCP (=Valproate) so better for birth control , and vice versa.
Most of AEDs are teratogenic; Category D
Therapy not stopped; uncontrolled seizure is risky to fetus & mother. Give lowest effective dose.
Avoid phenytoin, valproate and barbiturates (use Lamotrigine)
Most AEDs cause folate deficiency …. Folic acid (prior to or early in conception)
Most AEDs are competitive inhibitors of vit. K-dependent clotting factor: Vit. K to mother 10 days before labor & to newborn.
Most except carbamazepine and valproate are present in breast milk. Lamotrigine is safe on infants.
Status Epilepticus:
WHAT? Seizures lasting for >30min, OR repeated seizures without intervening consciousness.
Things to be done:
Bedside glucose, the following tests can be done once Rx has started: lab glucose, ABG, U&E, Ca2+, FBC, ECG.
Consider anticonvulsant levels, toxicology screen, LP, cultures, EEG, CT, CO level.
Pulse oximetry, cardiac monitor.
How to treat?

THE END

Inhibidores del citocromo P450
“ERIc, que es un chico muy inhibido, está enCIMa de RITa ACOjonado y Kieto chupando un POMELO, que es ÁCIDO.”
ERItromicina
EnCIMetidina
RITonavir
ACO - Anticonceptivos orales
Ketoconazol (y otros azoles)
Pomelo
ÁCIDO valproico
Fuente: tutor de la academia